ChemDrawRevvity 中国区官方代理
发布时间:2016-06-12 13: 47: 24
接触过结构化学的人都知道分子的结构决定其性质,在实际实验中时常会因为中间体寿命过短或者是混合物难以分离使得观测到的分子结构不稳定,此时就需要借助计算化学来帮忙预测,即构型优化。所谓构型优化就是找该体系的最小点或鞍点,优化分子构型是研究中的难点,本节ChemDraw教程将指导使用Chem 3D软件轻松优化分子构型的两种方法。
所有计算化学研究分子性质均是从优化分子结构开始,在自然情况下分子主要以能量最低的子女格式存在,所以低能的分子结构具有代表性,这样也才能保证得到的计算结构有意义,Chem 3D软件使用MM2分子力学和Gamess量子两种方法来优化构型。
使用Chem 3D软件MM2分子力学优化构型的操作步骤是绘制出化学结构之后,依次选择Calculations/MM2/Minimize Energy(最小化化学能)命令,如下图所示:
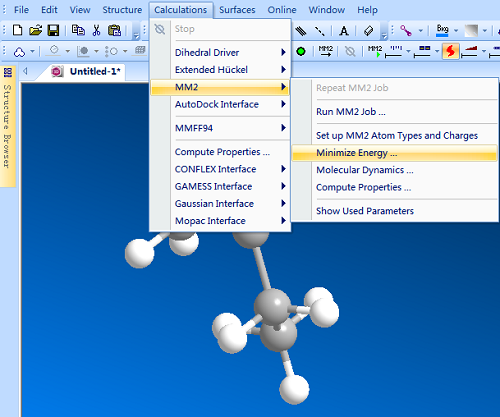
接着会弹出Minimize Energy对话框,“Display Every nth Iter用于显示每轮迭代信息”、“Copy Measurements to Output Bo用于控制输出每轮结构参数”、“Minimum RMS是构型收敛标准”。另外注意一下,MM2分子力学方法计算量小,适合于大体系有机分子的构型优化。
Gamess量子化学软件包进行构型优化的原理是Chem 3D根据初始分子模型计算能量和梯度,然后决定写一部结构调整的方向的步长,根据各原子受力情况和位移大小判断是否收敛,若没有则继续重复上面的过程直到力和位移的变化均达到收敛标准。在Chem 3D软件中的操作方法是:绘制出分子结构之后,依次点击Calculations/GAMESS Interface/Minimize,随后会弹出如下图所示的GAMESS对话框。
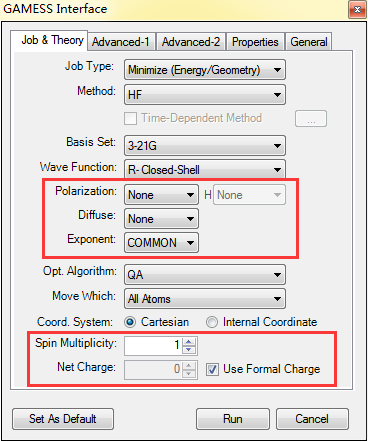
对话框Job&Thery选项卡的Method选项可以更改计算方法,Basis Set是基组类型,Wave Function是波函数类型,通过Polarization、Diffuse和Exponent可以添加或弥散基函数,Opt.Algorithm用于修改构型优化方法,最后的两个选项Spin Multiplicity和Net Charge指的是体系电荷和自选多重度。在Advanced-1选项卡中,绘制者可以更改自洽场迭代参数控制、溶剂效应及其模型、MO初始猜测类型、点群及其主轴。GAMESS Interface所计算性质包括:偶极矩、电子密度、静电势、动能、Lowdin电荷和布居数、Mulliken电荷和布居数、势能和总能量。
以上就是使用Chem 3D软件优化分子构型的两种方法介绍,更多ChemOffice组件使用技巧请点击Chem 3D中如何计算化学数据?
展开阅读全文
︾



